GMP制药洁净车间设计施工



公司介绍 网站公告  新闻中心 新闻中心
产品分类联系方式
站内搜索 荣誉资质
友情链接 |
GMP制药洁净车间设计施工 《药品生产质量管理规范(2010年修订)》
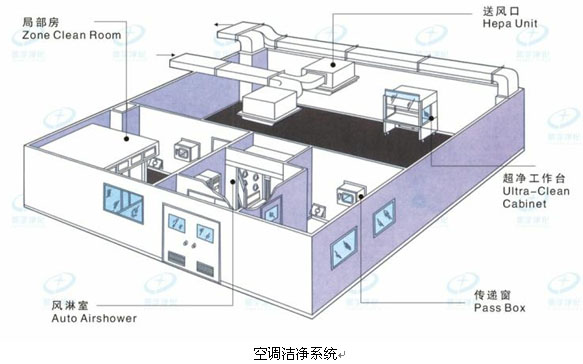
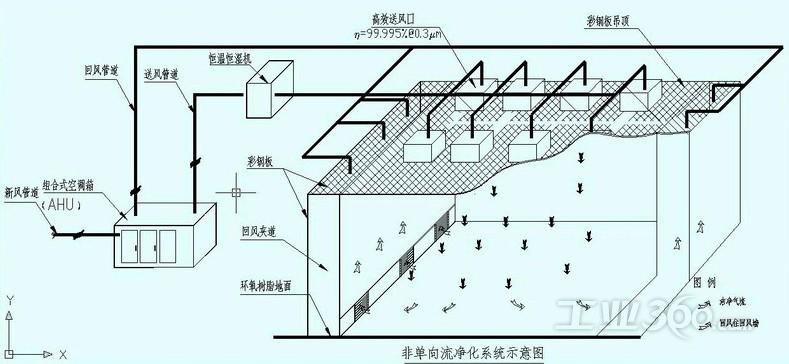
无菌制剂 第四条 无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。 第五条 无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。 第六条 物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。 第八条 洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。 第九条 无菌药品生产所需的洁净区可分为以下4个级别: A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。 B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。 C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:
第十一条 应当对微生物进行动态监测,评估无菌生产的微生物状况。监测方法有沉降菌法、定量空气浮游菌采样法和表面取样法(如棉签擦拭法和接触碟法)等。动态取样应当避免对洁净区造成不良影响。成品批记录的审核应当包括环境监测的结果。 洁净区微生物监测的动态标准(1)如下:
第三十一条 气锁间两侧的门不得同时打开。可采用连锁系统或光学或(和)声学的报警系统防止两侧的门同时打开。 第三十四条 应设送风机组故障的报警系统。应当在压差十分重要的相邻级别区之间安装压差表。压差数据应当定期记录或者归入有关文挡中。 第三十五条 轧盖会产生大量微粒,应当设置单独的轧盖区域并设置适当的抽风装置。
●A~D级GMP医药工业洁净厂房设计、建造及维护保养; ●N9级~N5级微电子无尘车间、光电净化车间设计施工; ●Ⅰ~Ⅳ级SC食品工业洁净用房设计、建造; ●Ⅰ~Ⅳ级医院洁净手术部、ICU、、供应室设计、安装及维护保养; ●30万级~100级医疗器械净化车间、消毒用品车间设计施工; ●生物安全、动物实验室及其他行业通风净化系统设计、建造及维护保养; 江西全立森洁净技术有限公司 何工:130 2728 7520
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||